Introduction to biology (for computer scientists)
Learning outcomes
At the end of this chapter, students will be able to:
Enumerate the key biological molecules/strings that are typically analyzed in bioinformatics (DNA, RNA, protein)
Describe how DNA, RNA, and protein are related to each other in the context of the central dogma of molecular biology
Define basic terminology such as codon, splicing, exon/intron, 5'-3', etc.
Reverse complement strings of DNA
Identify open reading frames within a string of DNA
Assess the impact of mutations within genes/open reading frames on an organism
Describe the implications of the ambiguity in the genetic code
Describe some of the technologies used to sequence DNA
Describe how DNA and protein sequences are encoded computationally
Quick introduction to biology and biological sequences
Many biological objects can be interpreted as strings. DNA, for example, is usually encoded as a string built from a 4-letter alphabet {A, C, T, G}, corresponding to the four nucleotides forming the DNA double-helix. Proteins can also be seen as strings built from a 20 letter alphabet, corresponding to the 20 different amino-acids commonly used by living organisms. These lecture notes primarily focus on concepts related to the analysis of such biological strings. First, we'll introduce the context within which these strings exist in life. This quick introduction to molecular biology is neither comprehensive, nor fully accurate, rather it is just meant to quickly introduce people without prior biology knowledge to some of the key terms in the field. You will undoubtedly encounter other terms you do not know in both these lecture notes and in other contexts, and you should spend some time searching some of the terms on the internet, or reading various introductory textbooks (the Cartoon Guide to Genetics by Larry Gonick – ISBN 978-0062730992 – is a surprisingly good first read).
The central dogma of molecular biology
The central dogma of molecular biology attempts to explain the flow of information in self-replicating biological systems. In this model, genetic information is encoded in DNA. A molecular process, called transcription, copies the genetic information into (nearly) identical RNA molecules, which are then converted into proteins through a process called translation. Proteins are the biological objects that perform all the functions of the cell, including the actual transcription and translation processes, as well as the DNA replication function. This general model can broadly explain how living organisms operate, though in recent years scientists have uncovered substantially more complex features of the mechanisms of life, such as the fact that genetic information can be transmitted not just through DNA alone but also through 'annotations' along the DNA (e.g., methylation status) or even through the way in which the DNA is physically organized within a cell.
The terms transcription and translation are well ingrained in the vocabulary of biologists and are taught as part of Biology 101, thus, it may come as a surprise that these terms were actually proposed by John von Neumann, a pioneer of computer science. Von Neumann proposed this two stage process, comprising a blind replication step followed by the interpretation/execution of the instructions, as a way to achieve self-replicating automata. It is remarkable that his intellectual model turned out to so closely model the actual operating principles of life. To give credit to where credit is due, however, von Neumann's model arose from philosophical discussions that were taking place at the time between mathematicians, biologists, and philosophers. This is just one of many examples that demonstrate the value of cross-disciplinary dialogue in promoting scientific progress.
DNA and self-replication
DNA has a particular structure that is essential for its role as a 'magnetic tape' storing our genetic information. DNA is a polymer, a molecule composed by the chaining of four different small molecules called nucleotides or bases. These four nucleotides (adenine – A, guanine – G, cytosine – C, thymine – T) can be chained in an arbitrary order along a sugar backbone, allowing the encoding of essentially arbitrary information. The particular sequence of nucleotides in a strand of DNA is commonly represented by a string of letters comprised of the first letters of the nucleotides (A, G, C, T). Usually, the chromosomes of organisms consist of two such chains (or strands) that are twisted around each other in a double-helix. The two strands are held together by chemical bonds between 'complementary' nucleotides. Specifically, the nucleotides fall into two classes: purines (A, G) and pyrimidines (C, T) based on their chemical composition (purines are larger and contain two rings while pyrimidines only contain one ring). Their chemical structure allows the specific pairing of the purine adenine with the pyrimidine thymine (A-T) and of the purine guanine with the pyrimidine cytosine (G-C), and these bonds hold together the double-helix structure.
A by-product of this paired structure of DNA is redundancy in the genetic information encoded in an organism's chromosomes. Specifically, the sequence of one strand can be determined from the sequence of the other by simply reconstructing the complementary structure of its bases. This property is key for the ability to copy the genetic material of one cell and to distribute these copies to its daughter cells during cell division. To perform a copy, the double helix is unwound, and a chemical reaction synthesizes a sister strand for each of the two unwound strands, leading to the creation of two double helices, each an identical copy of the original.
It is important to note that the process of replication always occurs in the same direction along the DNA strand—a direction termed 5' to 3' due to the chemical nomenclature for the ends of the DNA polymer (for more details check out a biochemistry manual or online resources). By convention, the string of letters representing a DNA molecule is always written in the direction 5' to 3'. Converting the sequence of one strand to that of the other, thus requires one to both change all letters to their complement, and to also invert their order, a procedure called reverse complementation. For example, see the two strands represented below and the correct representation of the DNA composition of the second strand.
strand 1: 5' – A C A G T G C A T G C C T G A – 3'
strand 2: 3' – T G T C A C G T A C G G A C T – 5'
string representation of strand 2: 5' – T C A G G C A T G C A C T G T – 3'
RNA—it's just another type of DNA
Ok, this is just a computer scientist's view and biologists will strongly disagree with this statement. RNA is a molecule that is very similar to DNA, where the thymine nucleotide is changed to uracil (U). Unsurprisingly U and A form a complementary bond, matching the situation found in DNA. Where RNA differs from DNA is in it's structure and function within the cell. Above we described a situation where RNA is simply a 'crib sheet' used to copy part of the DNA as a step towards generating a protein. In this setting RNA lives as a single stranded molecule. An important feature of single stranded RNA is that it can fold onto itself and form base pairings along the same strand, as seen below.

{kind=link}
This folded structure makes the RNA molecule rigid and allows it to be used as a structural element in other cellular objects. A good example is the ribosome—a complex of proteins and structural RNA that is the molecule which synthesizes proteins from the template provided by a gene (more on that below). The folded RNA structure (also called secondary structure) also plays a role in gene expression, for example by stopping certain biochemical processes such as transcription or translation (sort of like a knot on a rope preventing it from going through a hole).
It is important to note that such self folding is not only restricted to RNA and that single stranded DNA molecules can also self-fold. Furthermore, as we'll discuss in a bit more detail below, most biological strings (including double-stranded DNA) can and do fold up in complex structures that ultimately affect their function in the cell.
Also note that RNA can also exist in a double-stranded form. Such a form is often used by viruses to encode their chromosomes, thus many organisms have developed mechanisms to destroy double-stranded RNA as a defense against invading viruses. Similar mechanisms can also be used on purpose to destroy RNA produced within the cell as a way to regulate the amount of RNA and/or protein produced in the cell. Such processes are part of the fascinating field of gene regulation.
Proteins—the sequence is just the beginning
Proteins are the cellular molecules that 'do stuff' inside the cell—they are generally responsible for the machinery of life. Proteins are, like DNA, polymer chains composed of smaller molecules called amino acids. Amino acids have the same backbone (with the generic chemical formula H2N-CH-R-COOH) which is decorated with a side-chain (the R in the earlier formula) which determines the type of amino acid (see below)

{kind=link}
The amino acids are chained together through chemical bonds between the extreme nitrogen and carbon atoms. There are 20 amino acids generally used in most organisms, thus, proteins can be viewed as strings of letters over a 20-letter alphabet.
Viewing proteins as strings of amino acids is, however, overly simplistic as these molecules owe their functionality to their three-dimensional folded structure (see below). The task of inferring this three-dimensional shape (also called tertiary structure) is the subject of the sub-field of protein folding, and is one of the big unsolved problems in bioinformatics (despite significant progress made in this space, e.g., through the use of deep learning, see AlphaFold).
Translation—how RNA becomes protein
As mentioned above, the central dogma of life posits that the information contained in DNA (strings over an alphabet of 4 letters) gets copied into RNA (also strings over an alphabet of 4 letters) and then converted into proteins (strings over an alphabet of 20 letters). For this translation to be possible, each amino acid must be encoded by a pattern of three or more nucleotides (42=16<20, 43=64>20). Biological systems use a genetic code of exactly three letters. Note that this code is redundant (multiple codes yield to a same amino acid, see below)
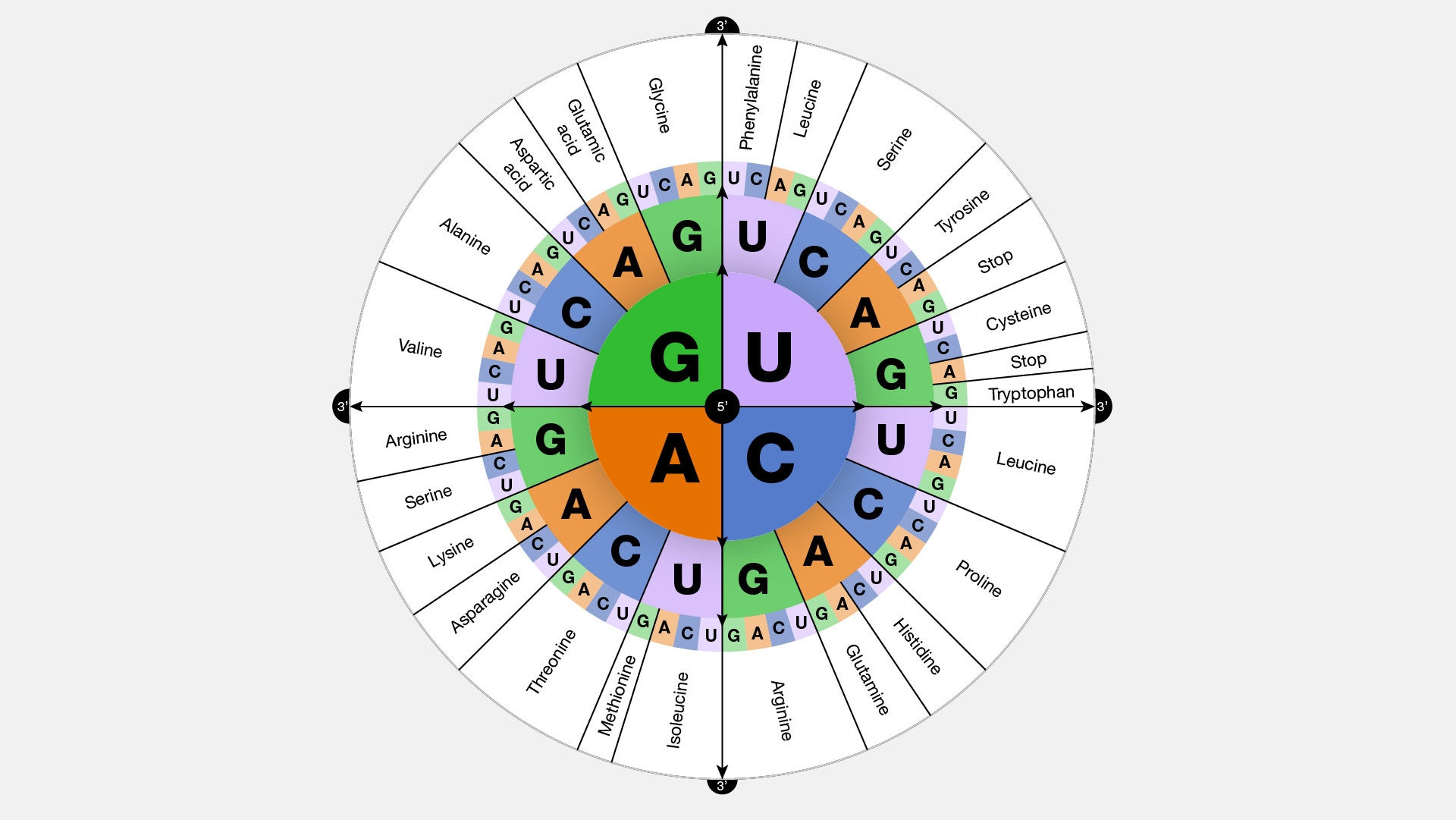
Within a chromosome only specific segments become proteins—these are called genes and are delimited by special signals in the DNA sequence. Simplistically, genes start with a start codon, usually AUG (or ATG in DNA) which also encodes the amino acid methionine. Genes end with one of three different stop codons (UAA, UAG, UGA). It is important to note that the start and stop codons must be in frame, i.e., the length of the string of DNA separating them must be a multiple of 3, as the cellular machinery (a protein-RNA complex called the ribosome) reads the RNA in groups of 3.
RNA splicing
In higher organisms the genes are often broken up in the chromosome into a collection of exons which are separated by introns. During the transcription into an RNA molecule, the exons are spliced together and the introns are discarded, and the translation into protein proceeds from the spliced transcript. It is possible that the same set of exons and introns can produce different transcripts (and therefore different proteins) through a process called alternative splicing, process which splices together different combinations of the starting exons and introns. Through this process, the fairly small number of genes found in the human genome (~26,000) yields the tremendous complexity embodied in our organism (many simpler organisms have more genes than we do).

DNA sequencing—how we 'read' DNA
The ability to read the DNA sequence of an organism's chromosomes has revolutionized modern biological research, and has inspired much of the research described in this course. At the same time, modern DNA sequencing technologies have emerged from a meaningful and deep collaboration between biologists, biochemists, engineers, and computer scientists. Future transformative developments in bioinformatics and other fields will only be achievable through similarly multi- and inter-disciplinary interactions. In other words: learn to understand and work with others!
Existing DNA sequencing technologies take advantage of the natural machinery that replicates DNA, which they modify so that they can 'spy' on the DNA as it is being synthesized. For example, Sanger sequencing, a technology that dominated the field for over 30 years (and which yielded Frederick Sanger, its inventor, a Nobel prize), uses modified nucleotides that block the DNA replication process. By carefully tuning the ratio of normal and modified nucleotides the biochemical reaction that replicates DNA ends up generating a population of DNA sequences of different lengths (imagine each of them stopped at a random location in the replication process). These sequences are then sorted by length within a gel and the DNA sequence is being reconstructed by reading the terminating base (which is usually tagged with a colored probe). This approach is a first generation technology, and is fairly low throughput—the most advanced instruments can only read about 384 sequences at the same time.
A new breed of sequencing technologies, the so called second generation technologies, have emerged in the early 2000s and use a slightly different approach to read DNA. Generally, millions of single stranded DNA molecules are immobilized on a surface, then an imaging device 'spies' on the DNA replication process as it proceeds simultaneously along all these molecules. The technologies differ primarily through the way in which the incorporation of new nucleotides is detected, usually through the emission of light. This approach achieves much higher throughput (tens to hundreds of millions of sequences are being read simultaneously) but read shorter segments than the Sanger technology (hundreds as opposed to thousands of letters).
More recently, third generation technologies have emerged that are able to sequence longer DNA segments (up to tens of thousands of letters) as well as to avoid the need for DNA amplification technologies. DNA amplification is essential in the first and second generation technologies in order to ensure sufficient signal is generated during the sequencing process, however amplification also introduces errors.
Here I will not go into more details about the specific technologies available today but encourage you to search online the following terms: Sanger sequencing, Roche/454, Illumina/Solexa, ABI Solid, Ion Torrent, Pacific Biosciences, Oxford Nanopore.
Representing sequences computationally
There are two major formats commonly used to represent biological sequences. The oldest and widest used is the FASTA format. It is an entirely ASCII text format that follows the pattern:
>seq1 some more information
ACCGGTAGCATAGA
CGGATAGACTTAGT
GCATT
>seq2 etc
...
Specifically, each sequence (either DNA or amino-acid) is prefixed by a one line header starting with a greater-than sign. The header then contains some sequence identifier (generally similar in syntax to what you would use as an identifier in a programming language—no white space, characters, digits, and some symbols allowed) and also some additional free-form information. The latter can be more precisely defined for specific applications. Each header is then followed by one or more rows of sequence information, separated by newlines. There are no specific requirements on the width of these rows, other than an unofficial standard keeping them at 60 characters (to fit on an old 80x24 CRT terminal). Of course, the last line of each record can and often is shorter.
The need to create a new file format arose when second generation sequencing technologies emerged. This format—FastQ—is also split into a collection of records, one for each sequence, and each of which uses up four rows as follows:
@seqid other info
ACCAGTACGTCCGTG
+seqid other info (optional)
!+30qr-130!@+-@
Just like the FASTA file, the first and third lines encode header information including a sequence identifier, prefixed by the 'at' sign (first line) and the plus sign (third line). The information on the second line is the actual DNA sequence, while the last line in the record encodes the quality information for each nucleotide. The latter is information provided by the sequencing instrument that describes the uncertainty in the specific call (essentially an estimate of the noise in the signal processing used to convert the signal into a DNA sequence).
The quality information merits a few additional qualifications. First, the quality concept was introduced in Sanger sequencing and is supposed to be the estimated probability of error. For example, a quality value of 20 indicates a 1 in 100 probability of error. Formally, the quality value q is related to the probability of error by the following formula:
Second, the letter string encoded in the FASTQ format is a translation of the actual quality values into printable ASCII characters. For example, an offset of 48 would convert the number 0 into the ASCII character '0' (the zeroth position in the ASCII table is the unprintable NUL character).
While the FASTQ format is quite widely used it has several major limitations. First, it is unclear why datasets commonly comprising tens of millions of sequences need to be encoded in a human readable format. Second, the header information is unnecessarily verbose (it can even be duplicated in the first and third lines of the record) and is often of roughly the same length as the sequence itself. Finally, the function translating integers into printable characters is not standardized nor obvious from the file itself. There are, in fact, several competing and incompatible formats that cannot be immediately distinguished from each other. A much better option would have been the development of a binary file format and associated utilities for viewing, editing, and converting this information.
Exercises
Write code that parses a FASTA file and outputs a tab-delimited file that contains the sequence IDs and corresponding sequence lengths for each of the records in this file.
Write code that parses a FASTQ file and outputs a tab-delimited file that contains the sequence IDs and corresponding sequence lengths for each of the records in this file.
Write code that parses a FASTQ file and changes the offset for the encoding of quality values. For example, the first printable character (corresponding to quality value 0) could be ASCII 33 (!) or ASCII 64 (@), or ASCII 65 (A) or ASCII 48 (0). Your code should accept as input the current offset for the file and the desired new offset, and output an error if it detects that the current file format does not match the offset indicated by the user.
Reverse complement the following DNA string: CAACAGGTCTATAAATTGGG
Which of the following mutations has the most significant impact on the protein sequence generated by the following DNA string? Assume the start codon is ATG and there is only one stop codon, TAA. The 'v' indicates that the letter above it was mutated to the letter below. If the letter below is '-', this indicates a deletion.
Last updated